R语言编程
20230405_基于R语言的MetaTX软件包绘制Meta gene图

Meta gene分析及常用软件:
在基因表达研究中,研究人员通常会将一组基因或转录本的表达模式相加或取平均值,以获得它们在特定区域(如启动子、非编码区、CDS区等)的平均表达情况。这种表达模式通常称为meta gene.通过绘制meta gene,可以更直观地了解这组基因或转录本的表达模式,从而深入探究基因调控机制和信号通路的调节。绘制Meta gene图的软件有很多种,以下是一些常用的软件:
- MetaPlotR:MetaPlotR是一种基于R语言的绘制Meta gene图的软件。它支持多种数据类型,包括基因表达、ChIP-seq、ATAC-seq和DNA甲基化等数据类型,同时还具有丰富的可视化选项,可以方便地自定义图表样式。
- ngs.plot:ngs.plot是一种基于Python语言的绘制Meta gene图的软件。它支持多种数据类型,包括RNA-seq、ChIP-seq、ATAC-seq和WGBS等数据类型,并且具有自动化流程,可以方便地生成高质量的Meta gene图。
- Guitar:Guitar是一种基于R语言的绘制Meta gene图的软件,它支持RNA-seq和ChIP-seq等数据类型,并且具有丰富的可视化选项和易用的用户界面,可以方便地生成高质量的Meta gene图。
- MetaGene:MetaGene是一种基于Java语言的绘制Meta gene图的软件,它支持多种数据类型,包括RNA-seq、ChIP-seq和ATAC-seq等数据类型,并且具有多种绘图模式和可视化选项,可以方便地满足不同研究需求。
- seqMINER:seqMINER是一种基于R语言的绘制Meta gene图的软件,它支持多种数据类型,包括RNA-seq、ChIP-seq和microarray等数据类型,并且具有多种可视化选项和高度可定制的绘图功能,可以方便地生成符合用户需求的Meta gene图。
- MetaTX: 是一款用于绘制meta gene的生物信息学软件,它可以帮助研究人员更好地理解一组基因或转录本的表达模式,从而深入探究基因调控机制和信号通路的调节。
Tribe测序方法中应用meta gene分析的意义
TRIBE (targets of RNA-binding proteins identified by editing)是一种用于识别RNA结合蛋白靶点的技术。该技术利用RNA编辑酶在RNA上进行的化学修饰(如A-to-I编辑),来识别RNA结合蛋白与RNA的结合位点。TRIBE技术不仅能够鉴定RNA结合蛋白的靶点,而且还能够揭示RNA结合蛋白在RNA上的结合偏好和特定区域的结合强度。对于TRIBE测序结果绘制meta gene图的意义在于,可以对RNA结合蛋白在RNA上的结合偏好和特定区域的结合强度进行可视化。具体地,通过将TRIBE测序数据与基因组注释信息进行比对,可以确定RNA结合蛋白在RNA上的结合区域,并在这些区域内绘制meta gene图,以反映RNA结合蛋白的结合偏好和结合强度。这有助于更深入地理解RNA结合蛋白的生物学功能,以及其在调控RNA生物学过程中的作用。另外,TRIBE测序结果绘制meta gene图还可以用于比较不同条件下RNA结合蛋白的结合偏好和特定区域的结合强度,从而揭示RNA结合蛋白在不同条件下的结合变化和相应的调控机制。这对于深入了解RNA结合蛋白在不同生理或病理状态下的功能和调控机制具有重要意义。
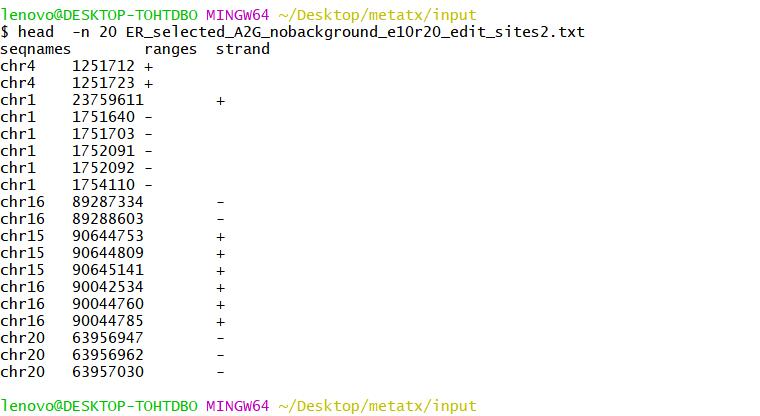
测试数据格式:
第一列为发生基因编辑事件的染色体编号,第二列为编辑事件在染色体上发生的具体坐标,第三列为基因所在的正负链。
MetaTX 包安装步骤:
#下载并安装4.2.3版本的R
https://mirrors.tuna.tsinghua.edu.cn/CRAN/bin/windows/base/
#下载metaTX的安装包的压缩文件
https://github.com/yue-wang-biomath/MetaTX.1.0
#本地安装metaTX
devtools::install_local("MetaTX.1.0-master.zip")
#测试是否安装成功
library(MetaTX)
install.packages("BiocManager")
library(BiocManager)
BiocManager::install("cowplot")
library(cowplot)
BiocManager::install("devtools")
install.packages("devtools")
library(devtools)
library(MetaTX)
#BiocManager::install(c("GenomicAlignments", "GenomicRanges", "GenomicFeatures", "ggplot2", "TxDb.Hsapiens.UCSC.hg19.knownGene", "cowplot"))
BiocManager::install(c("TxDb.Hsapiens.UCSC.hg38.knownGene"))
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
remove.packages(c("curl","httr"))
install.packages(c("curl", "httr"))
Meta gene图绘制:
txdb <- TxDb.Hsapiens.UCSC.hg38.knownGene
#$$$$$$$$$$$$$$$$$$$$
input <- read.table('./input/ER_selected_A2G_nobackground_e10r20_edit_sites2.txt',header = TRUE)
head(input)
seqnames <- input$seqnames
starts <- input$ranges
ends <- starts + 0
strands <- input$strand
gr <- GRanges(seqnames, IRanges(starts,ends), strand = strands)
total_sum = length(seqnames)
total_sum
ER_selected_A2G_nobackground_e10r20_edit_sites2 <- remapCoord(features = gr[1:940], txdb = txdb)
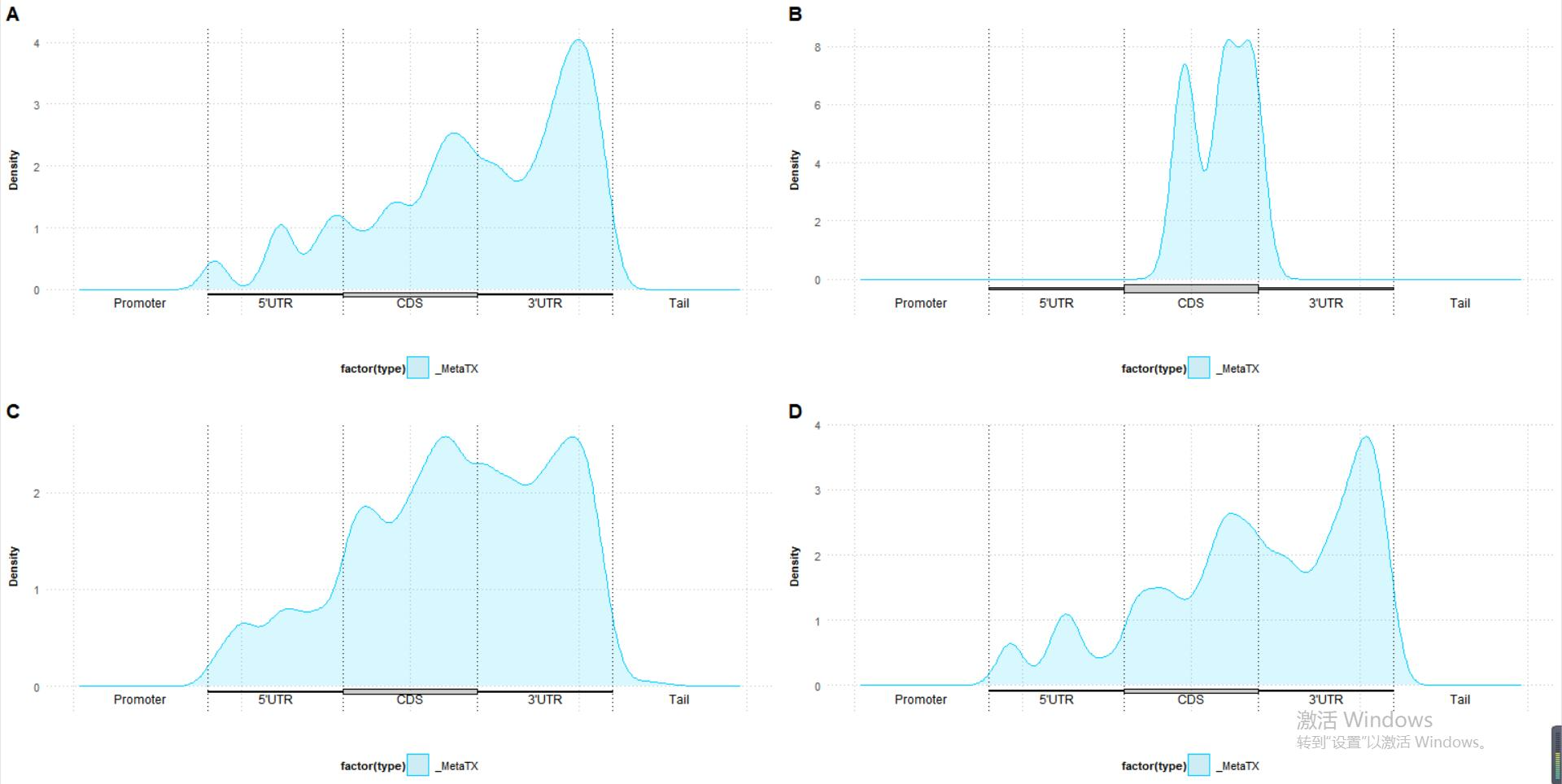
p1 <- metaTXplot(ER_selected_A2G_nobackground_e10r20_edit_sites2)
#$$$$$$$$$$$$$$$$$$$$
#$$$$$$$$$$$$$$$$$$$$
input <- read.table('./input/lin28A_A2G_nobackground_e10r20_edit_sites2.txt',header = TRUE)
head(input)
seqnames <- input$seqnames
starts <- input$ranges
ends <- starts + 0
strands <- input$strand
gr <- GRanges(seqnames, IRanges(starts,ends), strand = strands)
total_sum = length(seqnames)
total_sum
lin28A_A2G_nobackground_e10r20_edit_sites2 <- remapCoord(features = gr[1:64900], txdb = txdb)
p2 <- metaTXplot(lin28A_A2G_nobackground_e10r20_edit_sites2)
#$$$$$$$$$$$$$$$$$$$$
#$$$$$$$$$$$$$$$$$$$$
input <- read.table('./input/LIN28Atotal__A2G_nobackground_e10r20_edit_sites2.txt',header = TRUE)
head(input)
seqnames <- input$seqnames
starts <- input$ranges
ends <- starts + 0
strands <- input$strand
gr <- GRanges(seqnames, IRanges(starts,ends), strand = strands)
total_sum = length(seqnames)
total_sum
LIN28Atotal__A2G_nobackground_e10r20_edit_sites2 <- remapCoord(features = gr[23:6700], txdb = txdb)
p3 <- metaTXplot(LIN28Atotal__A2G_nobackground_e10r20_edit_sites2)
#$$$$$$$$$$$$$$$$$$$$
#$$$$$$$$$$$$$$$$$$$$
input <- read.table('./input/Cytosol_selected_A2G_nobackground_e10r20_edit_sites2.txt',header = TRUE)
head(input)
seqnames <- input$seqnames
starts <- input$ranges
ends <- starts + 0
strands <- input$strand
gr <- GRanges(seqnames, IRanges(starts,ends), strand = strands)
total_sum = length(seqnames)
total_sum
Cytosol_selected_A2G_nobackground_e10r20_edit_sites2 <- remapCoord(features = gr[4:1200], txdb = txdb)
p4 <- metaTXplot(Cytosol_selected_A2G_nobackground_e10r20_edit_sites2)
plot_grid(p1,p2,p3,p4,labels = LETTERS)
txdb <- TxDb.Hsapiens.UCSC.hg38.knownGene
#$$$$$$$$$$$$$$$$$$$$
input <- read.table('./input/ER_selected_A2G_nobackground_e10r20_edit_sites2.txt',header = TRUE)
head(input)
seqnames <- input$seqnames
starts <- input$ranges
ends <- starts + 0
strands <- input$strand
gr <- GRanges(seqnames, IRanges(starts,ends), strand = strands)
total_sum = length(seqnames)
total_sum
ER_selected_A2G_nobackground_e10r20_edit_sites2 <- remapCoord(features = gr[1:940], txdb = txdb)
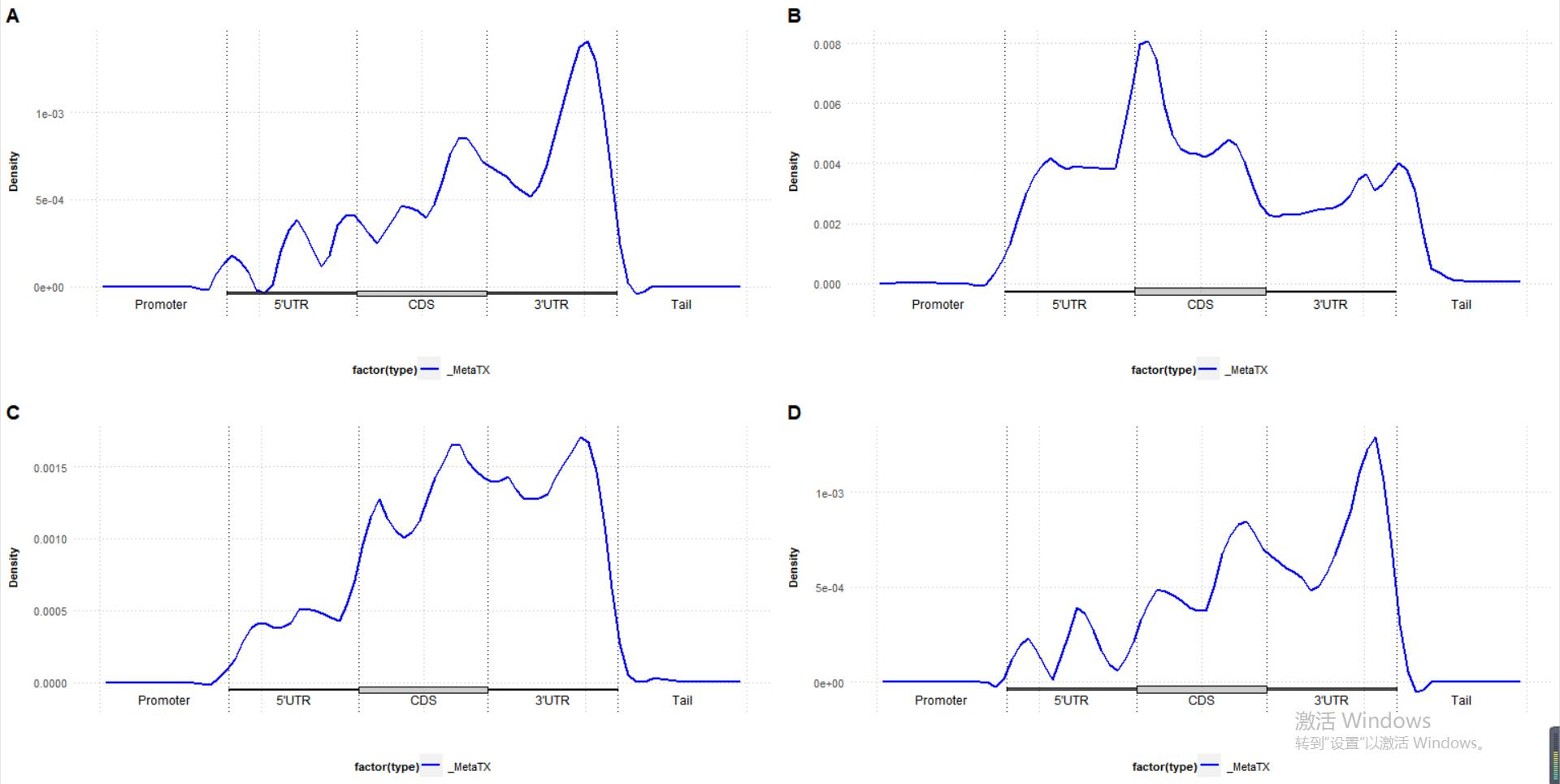
p11 <- metaTXplot(ER_selected_A2G_nobackground_e10r20_edit_sites2, type = 'absolute')
#$$$$$$$$$$$$$$$$$$$$
#$$$$$$$$$$$$$$$$$$$$
input <- read.table('./input/lin28A_A2G_nobackground_e10r20_edit_sites2.txt',header = TRUE)
head(input)
seqnames <- input$seqnames
starts <- input$ranges
ends <- starts + 0
strands <- input$strand
gr <- GRanges(seqnames, IRanges(starts,ends), strand = strands)
total_sum = length(seqnames)
total_sum
lin28A_A2G_nobackground_e10r20_edit_sites2 <- remapCoord(features = gr[1:64900], txdb = txdb)
p22 <- metaTXplot(lin28A_A2G_nobackground_e10r20_edit_sites2, type = 'absolute')
#$$$$$$$$$$$$$$$$$$$$
#$$$$$$$$$$$$$$$$$$$$
input <- read.table('./input/LIN28Atotal__A2G_nobackground_e10r20_edit_sites2.txt',header = TRUE)
head(input)
seqnames <- input$seqnames
starts <- input$ranges
ends <- starts + 0
strands <- input$strand
gr <- GRanges(seqnames, IRanges(starts,ends), strand = strands)
total_sum = length(seqnames)
total_sum
LIN28Atotal__A2G_nobackground_e10r20_edit_sites2 <- remapCoord(features = gr[23:6700], txdb = txdb)
p33 <- metaTXplot(LIN28Atotal__A2G_nobackground_e10r20_edit_sites2, type = 'absolute')
#$$$$$$$$$$$$$$$$$$$$
#$$$$$$$$$$$$$$$$$$$$
input <- read.table('./input/Cytosol_selected_A2G_nobackground_e10r20_edit_sites2.txt',header = TRUE)
head(input)
seqnames <- input$seqnames
starts <- input$ranges
ends <- starts + 0
strands <- input$strand
gr <- GRanges(seqnames, IRanges(starts,ends), strand = strands)
total_sum = length(seqnames)
total_sum
Cytosol_selected_A2G_nobackground_e10r20_edit_sites2 <- remapCoord(features = gr[4:1200], txdb = txdb)
p44 <- metaTXplot(Cytosol_selected_A2G_nobackground_e10r20_edit_sites2, type = 'absolute')
plot_grid(p11,p22,p33,p44,labels = LETTERS)


最新文章
推荐阅读
网站浏览
