
染色体共线性分析:
染色体共线性分析是一种比较两个或多个物种的染色体序列的相似性和差异性的方法。在染色体共线性分析中,研究人员可以确定染色体之间的共线区域和非共线区域,以及它们之间的结构和序列差异。这些信息可以提供有关物种之间遗传变异的详细信息,包括基因的位置、基因家族的扩张和缩小,以及整个染色体的演化历史。JCVI软件是目前使用最广的用来做染色体共线性分析的工具,但是无法对两个物种染色体间结构变异的数量进行定量统计。为了解决这一问题,我们感谢北卡罗来纳大学的阗瑞同学帮我们编写了一个脚本(structural_variation_identify.py),用于定量统计不同染色体间的移位、同一染色体内部的移位,以及染色体片段的倒置。这一方法为深入研究和理解物种染色体结构变异提供了有效的定量工具。
JCVI软件进行共线性分析:
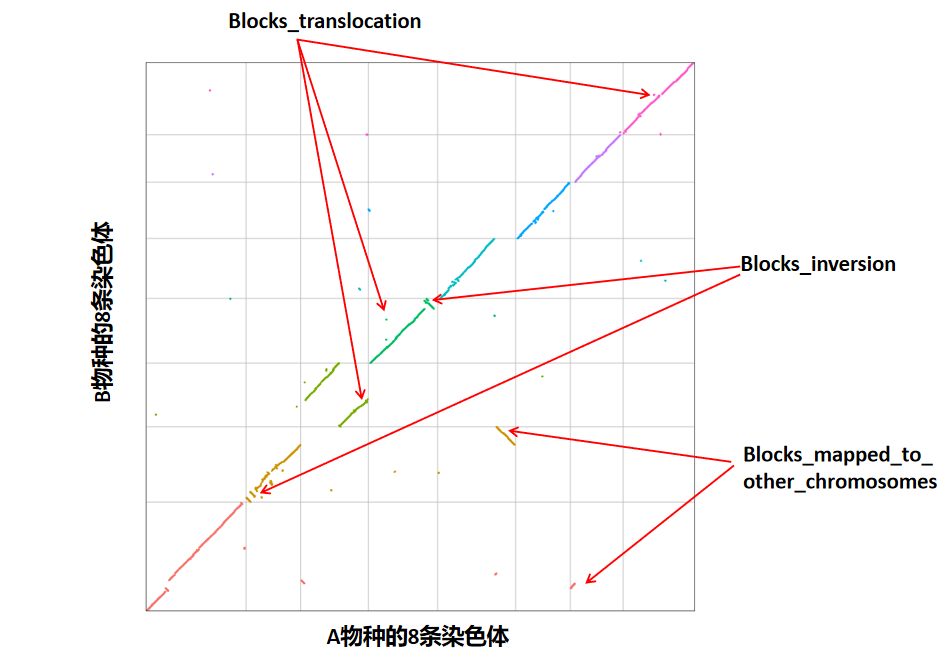
在本研究中,我们通过JCVI软件对比和鉴定两个物种染色体间的结构变异,旨在揭示它们染色体结构的相似程度。首先,我们将两个物种的染色体序列切割成等长的10,000个片段。接着,利用jcvi软件对这些切割后的片段进行相似性比较,并绘制共线性散点图。该散点图直观地展示了两个物种染色体之间的相似程度;共线性越高,染色体结构相似度越高。图中每个点代表一个切割的片段。
#JCVI软件运行命令docker run -v /var/run/docker.sock:/var/run/docker.sock -v `pwd`:/data -w /data test_tools:47.0_genome_compare_revised python /usr/bin/genome_comepare.py species_A_genome.fasta species_B_genome.fasta
JCVI软件输出结果一:
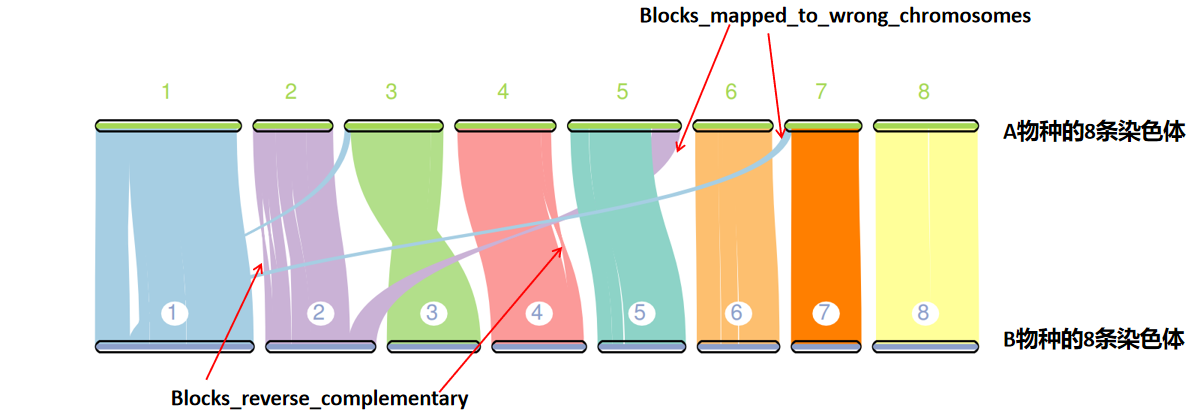
JCVI软件输出结果二:
JCVI软件输出结果三(collinearity_identify_result.txt):
这份文件揭示了两个物种染色体中具有相似性的区域。以第一行数据为例,它表明A物种的1号染色体上,从第974个片段至第1755个片段的序列与B物种的1号染色体上,从第1154个片段至第17967个片段的序列之间存在共线性。
chr1 part974 974 part1755 1755 CHR1 part1154 1154 part1967 1967 797 + chr1 part532 532 part964 964 CHR1 part691 691 part1122 1122 432 + chr1 part423 423 part526 526 CHR1 part561 561 part661 661 102 + chr1 part12 12 part356 356 CHR1 part2 2 part355 355 349 + chr1 part361 361 part409 409 CHR1 part363 363 part414 414 50 - chr1 part1784 1784 part1805 1805 CHR1 part1129 1129 part1153 1153 23 + chr1 part182 182 part186 186 CHR3 part3564 3564 part3572 3572 6 - chr1 part1533 1533 part1538 1538 CHR4 part5672 5672 part5677 5677 6 - chr1 part1210 1210 part1218 1218 CHR7 part7929 7929 part7942 7942 11 - chr1 part1161 1161 part1168 1168 CHR8 part9455 9455 part9459 9459 6 + chr2 part2340 2340 part2469 2469 CHR2 part2587 2587 part2705 2705 124 + chr2 part1828 1828 part1904 1904 CHR2 part1983 1983 part2060 2060 77 - chr2 part2288 2288 part2331 2331 CHR2 part2545 2545 part2586 2586 42 + chr2 part2112 2112 part2242 2242 CHR2 part2376 2376 part2524 2524 139 + chr2 part2477 2477 part2488 2488 CHR2 part2547 2547 part2557 2557 11 + chr2 part2495 2495 part2807 2807 CHR2 part2710 2710 part3019 3019 311 + chr2 part2098 2098 part2107 2107 CHR2 part2061 2061 part2074 2074 11 - chr2 part1986 1986 part2087 2087 CHR2 part2174 2174 part2283 2283 105 + chr2 part1894 1894 part1980 1980 CHR2 part2076 2076 part2170 2170 90 - chr2 part2285 2285 part2297 2297 CHR2 part2289 2289 part2306 2306 15 + chr2 part2254 2254 part2284 2284 CHR2 part2315 2315 part2370 2370 41 + chr2 part2114 2114 part2121 2121 CHR2 part2348 2348 part2355 2355 8 + chr2 part2090 2090 part2096 2096 CHR2 part2309 2309 part2321 2321 9 + chr2 part2737 2737 part2740 2740 CHR3 part3713 3713 part3716 3716 4 + chr3 part2822 2822 part2878 2878 CHR1 part502 502 part560 560 57 - chr3 part3361 3361 part3369 3369 CHR2 part2189 2189 part2199 2199 9 - chr3 part2896 2896 part3507 3507 CHR3 part3833 3833 part4503 4503 640 + chr3 part3508 3508 part3876 3876 CHR3 part3350 3350 part3711 3711 365 + chr3 part3903 3903 part4034 4034 CHR3 part3719 3719 part3852 3852 132 + chr3 part3274 3274 part3284 3284 CHR3 part4365 4365 part4395 4395 18 + chr3 part2883 2883 part2887 2887 CHR3 part4152 4152 part4156 4156 5 - chr3 part3867 3867 part3894 3894 CHR5 part5837 5837 part5862 5862 26 - chr3 part4002 4002 part4028 4028 CHR8 part8651 8651 part8659 8659 15 - chr4 part4515 4515 part4526 4526 CHR2 part2526 2526 part2537 2537 12 + chr4 part4078 4078 part5059 5059 CHR4 part4505 4505 part5489 5489 983 + chr4 part4366 4366 part4371 4371 CHR4 part5293 5293 part5298 5298 6 - chr4 part5126 5126 part5228 5228 CHR4 part5491 5491 part5594 5594 103 - chr4 part5098 5098 part5126 5126 CHR4 part5642 5642 part5671 5671 29 - chr4 part5060 5060 part5099 5099 CHR4 part5595 5595 part5639 5639 42 - chr4 part4362 4362 part4366 4366 CHR4 part4929 4929 part4932 4932 4 - chr4 part4041 4041 part4066 4066 CHR6 part7267 7267 part7298 7298 28 - chr5 part6342 6342 part6365 6365 CHR1 part662 662 part686 686 24 + chr5 part6367 6367 part6697 6697 CHR2 part3020 3020 part3347 3347 329 - chr5 part5313 5313 part5319 5319 CHR2 part2508 2508 part2514 2514 7 + chr5 part6328 6328 part6337 6337 CHR4 part5360 5360 part5369 5369 10 - chr5 part5619 5619 part6325 6325 CHR5 part6022 6022 part6761 6761 723 + chr5 part5574 5574 part5631 5631 CHR5 part5912 5912 part5977 5977 61 + chr5 part5494 5494 part5542 5542 CHR5 part5864 5864 part5911 5911 48 + chr5 part5339 5339 part5501 5501 CHR5 part5680 5680 part5835 5835 159 + chr5 part5562 5562 part5567 5567 CHR5 part5978 5978 part5983 5983 6 + chr5 part5664 5664 part5679 5679 CHR5 part5991 5991 part6015 6015 20 - chr6 part7197 7197 part7204 7204 CHR3 part4261 4261 part4265 4265 6 + chr6 part6749 6749 part7012 7012 CHR6 part6768 6768 part7039 7039 267 + chr6 part7036 7036 part7068 7068 CHR6 part7075 7075 part7104 7104 31 + chr6 part7232 7232 part7687 7687 CHR6 part7299 7299 part7779 7779 468 + chr6 part7089 7089 part7218 7218 CHR6 part7106 7106 part7259 7259 141 + chr6 part7070 7070 part7085 7085 CHR6 part7047 7047 part7068 7068 18 + chr6 part7396 7396 part7399 7399 CHR6 part7264 7264 part7267 7267 4 - chr7 part7713 7713 part7789 7789 CHR1 part415 415 part501 501 81 + chr7 part7799 7799 part8616 8616 CHR7 part7793 7793 part8650 8650 837 + chr7 part8606 8606 part8616 8616 CHR8 part8690 8690 part8700 8700 11 + chr8 part9431 9431 part9436 9436 CHR5 part5999 5999 part6004 6004 6 - chr8 part8986 8986 part8993 8993 CHR5 part6357 6357 part6365 6365 8 - chr8 part8697 8697 part9329 9329 CHR8 part8702 8702 part9364 9364 647 + chr8 part9371 9371 part9952 9952 CHR8 part9388 9388 part9968 9968 581 + chr8 part9219 9219 part9236 9236 CHR8 part9368 9368 part9384 9384 17 + chr8 part8673 8673 part8681 8681 CHR8 part8678 8678 part8687 8687 9 + chr8 part9341 9341 part9348 9348 CHR8 part8652 8652 part8670 8670 12 -
通过structural_variation_identify.py脚本定量统计两个物种的染色体之间结构变异的数量:
python structural_variation_identify.py collinearity_identify_result.txt #collinearity_identify_result.txt是上面jcvi生成的第三个结果#运行结果如下所示chr1<--->chr1 Mapped_to_other_chrns_num: 4 Inversion_num: 1 Translocation_num: 1chr2<--->chr2 Mapped_to_other_chrns_num: 1 Inversion_num: 3 Translocation_num: 8chr3<--->chr3 Mapped_to_other_chrns_num: 4 Inversion_num: 1 Translocation_num: 3chr4<--->chr4 Mapped_to_other_chrns_num: 2 Inversion_num: 5 Translocation_num: 4chr5<--->chr5 Mapped_to_other_chrns_num: 4 Inversion_num: 1 Translocation_num: 2chr6<--->chr6 Mapped_to_other_chrns_num: 1 Inversion_num: 1 Translocation_num: 3chr7<--->chr7 Mapped_to_other_chrns_num: 2 Inversion_num: 0 Translocation_num: 0chr8<--->chr8 Mapped_to_other_chrns_num: 2 Inversion_num: 1 Translocation_num: 2Blocks_mapped_to_other_chrns: 20 Blocks_inversion: 13 Blocks_translocation: 23 Total_errors: 56
上述结果表示共有20个片段被组装到其它染色体上,有13个片段需要反向互补,另外有23个片段被在染色体内发生了移位突变,最终共有56处结构变异。
============================
structural_variation_identify.py脚本内容:
import os
import sys
inputfile1 = sys.argv[1]
os.system('rm -rf error_summary && mkdir error_summary ')
os.chdir('./error_summary')
def remove_inner_blocks(file1) :
output = file1.strip().split(".txt")[0] + "-remove_inner_blocks.txt"
os.system('rm -rf %s'%(output))
aa = open(file1).read().strip().split("\n")
total_error = 0
i = 0
while i < len(aa) :
ele1 = aa[i]
num11 = ele1.strip().split("\t")[7]
num12 = ele1.strip().split("\t")[9]
tf = "false"
for ele2 in aa :
num21 = ele2.strip().split("\t")[7]
num22 = ele2.strip().split("\t")[9]
if int(num11) > int(num21) and int(num12) < int(num22) :
tf = "true"
else :
pass
if tf == "false" :
with open(output,"a+") as f :
f.write(aa[i].strip() + "\n")
else :
total_error = total_error + 1
i = i + 1
print(total_error)
return total_error
def wrong_position_error(test) :
#print(test)
test_sorted = sorted(test)
#print(test_sorted)
test_sorted_index = []
for ele in test_sorted :
test_sorted_index.append(test.index(ele) + 1)
#print(test_sorted_index)
#print(test_sorted_index[1:])
#print("====")
total_error = 0
ele1 = test_sorted_index[0]
if len(test_sorted_index) >= 2 :
if test_sorted_index[1] -1 == ele1 :
total_error = 0
ele1 = test_sorted_index[1]
else :
ele1 = test_sorted_index[1]
total_error = 1
for ele in test_sorted_index[2:] :
if ele - 1 == ele1 :
ele1 = ele
else :
if ele + 1 == ele1 :
ele1 = ele
else :
total_error += 1
ele1 = ele
else :
for ele in test_sorted_index[1:] :
if ele - 1 == ele1 :
ele1 = ele
else :
if ele + 1 == ele1 :
ele1 = ele
return total_error
def calculate(inputfile1) : #reference genome bed, query genome bed, anchors.simple file
os.system(' awk -F "\t" \'{print $0 > $1".raw.txt"}\' inputfile1 ')
chrn_files = os.popen('ls *.raw.txt').read().strip().split("\n")
print(chrn_files)
print("*******************")
for eles in chrn_files :
dic_tmp = {}
all_chr = os.popen('cut -f 6 %s | sort | uniq | sort -r -n | awk \'{print $2,$1}\' '%(eles)).read().strip().split("\n")
print("==============")
for e1 in all_chr :
print(e1)
block_num = int(os.popen('awk -F " " \'{ if ($6 == "%s") print }\' %s | awk \'{sum += $11 };END{print sum}\' '%(e1.strip(),eles)).read().strip() )
print(block_num)
dic_tmp[e1.strip()] = block_num
vlue1 = 0
for e2 in dic_tmp :
if dic_tmp[e2] >= vlue1 :
mapped_chr = e2
vlue1 = dic_tmp[e2]
vlue1 = 0
print(dic_tmp)
print(mapped_chr)
print("==============")
ref_chr = eles.strip().split(".raw.txt")[0]
fo = ref_chr + ".raw-rm-otherChr.txt"
##### calculate wrong blocks
os.system('awk \'{if ($1 == "%s" && $6 == "%s") print $0}\' %s > %s '%( ref_chr, mapped_chr, eles, fo ))
wrong_chrn_total = int(os.popen('awk \'{if ($1 == "%s" && $6 != "%s") print $0}\' %s | wc -l '%( ref_chr, mapped_chr, eles )).read().strip() )
##### calculate wrong blocks
strand_num = int(os.popen('cut -f 12 %s | sort | uniq | wc -l '%(fo)).read().strip() )
if strand_num > 1 :
positive_strand_length = int(os.popen(' awk \'{if ($12 == "+" ) print }\' %s | awk \'{aa = aa + $11 }END{print aa }\' '%(fo)).read() )
negative_strand_length = int(os.popen(' awk \'{if ($12 == "-" ) print }\' %s | awk \'{aa = aa + $11 }END{print aa }\' '%(fo)).read() )
if positive_strand_length >= negative_strand_length :
reverse_complementary_error = os.popen(' awk \'{if ($12 == "-" ) print }\' %s | wc -l '%(fo)).read()
else :
reverse_complementary_error = os.popen(' awk \'{if ($12 == "+" ) print }\' %s | wc -l '%(fo)).read()
else :
reverse_complementary_error = "0"
##### calculate wrong blocks
blocks_contained_in_other_blocks_totalnum = remove_inner_blocks(fo)
fo_2 = fo.strip().split(".txt")[0] + "-remove_inner_blocks.txt"
aa = os.popen('cut -f 8 %s '%(fo_2)).read().strip().split("\n")
bb = [int(x) for x in aa ]
total_wrong_position_error_tmp = wrong_position_error(bb)
total_wrong_position_error = int(blocks_contained_in_other_blocks_totalnum) + int(total_wrong_position_error_tmp)
output = file1.strip().split(".bed")[0] + "_-_" + file2.strip().split(".bed")[0] + "-error_summary.txt"
with open(output,"a+") as f :
f.write(ref_chr + "<--->" + mapped_chr.strip() + "\t" + "Mapped_to_other_chrns_num:" + "\t" + str(wrong_chrn_total) + "\t" + "Inversion_num:" + "\t" + str(reverse_complementary_error.strip()) + "\t" + "Translocation_num:" + "\t" + str(total_wrong_position_error) + "\n")
num1 = os.popen(' awk \'{aa = aa + $3} END {print aa}\' %s '%(output)).read().strip().split("\n")[0]
num2 = os.popen(' awk \'{aa = aa + $5} END {print aa}\' %s '%(output)).read().strip().split("\n")[0]
num3 = os.popen(' awk \'{aa = aa + $7} END {print aa}\' %s '%(output)).read().strip().split("\n")[0]
total = int(num1) + int(num2) + int(num3)
summary_content = "Blocks_mapped_to_other_chrns:" + "\t" + str(num1) + "\t" + "Blocks_inversion:" + "\t" + str(num2) + "\t" + "Blocks_translocation:" + "\t" + str(num3) + "\t" + "Total_errors:" + "\t" + str(total) + "\n"
with open(output,"a+") as f :
f.write("\n" + summary_content + "\n")
os.chdir('../')
os.system('cp ./%s/%s ./'%(output_fold,output))
calculate(inputfile1)


