附录:分析过程中使用到得代码
==================================
生物信息学

(1)基因组共线性是一种比较基因组学方法,用于比较不同物种或同一物种不同基因组中的基因组结构。它可以揭示出基因组中的保守基因区域和进化事件,例如基因家族的扩张、染色体重排、染色体片段重组和基因重复等。
(2)染色体共线性初步分析
nohup docker run -v /var/run/docker.sock:/var/run/docker.sock -v `pwd`:/data -w /data test_tools:47.0_genome_compare_revised python /usr/bin/genome_comepare.py mangguo-canu-hicpro-allhic-ok.fasta GCF_011075055-ok.fasta &cd mangguo-canu-hicpro-allhic-ok.GCF_011075055-ok
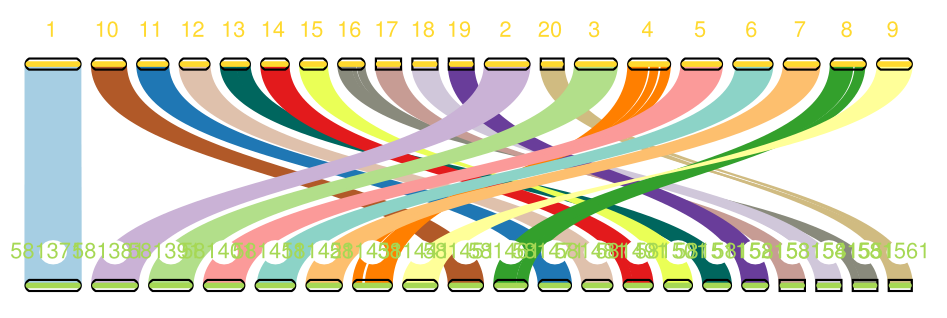
(3)根据上图结果,编辑seqids-two-line文件,对染色体进行重新排序,然后重新绘制基因组共线性结果。
vim seqids-two-line
docker run -v /var/run/docker.sock:/var/run/docker.sock -v `pwd`:/data -w /data genome_collinearity_plot_docker python -m jcvi.graphics.karyotype seqids-two-line layout-two-line
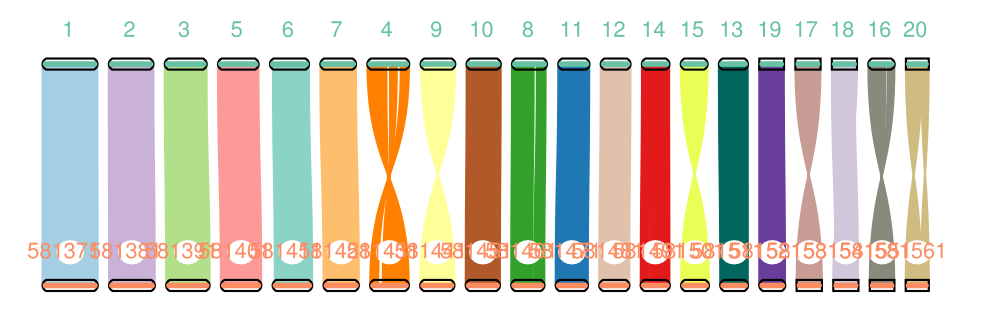
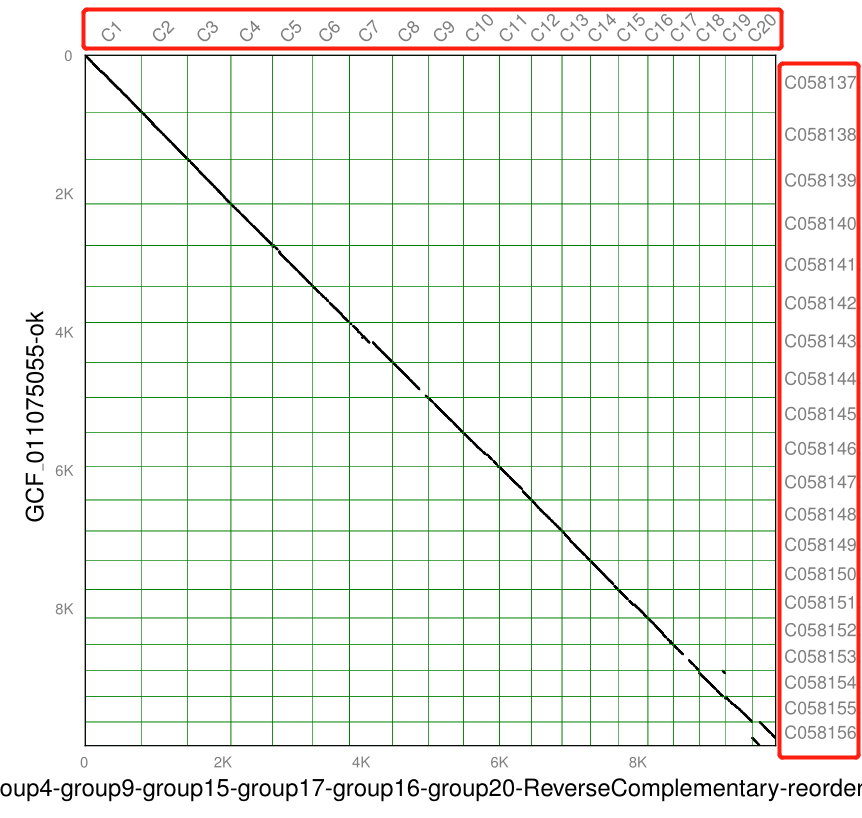
(4)将需要进行反向互补的染色体进行反向互补,并继续重新绘制基因组共线性结果。
将group4,group9,group15,group17,group16,group20这些染色体进行反向互补python chrom_ReverseComplementary.py mangguo-canu-hicpro-allhic-ok.fasta group4,group9,group15,group17,group16,group20cp mangguo-canu-hicpro-allhic-ok.fasta-group4-group9-group15-group17-group16-group20-ReversedComplementary_result/mangguo-canu-hicpro-allhic-ok-group4-group9-group15-group17-group16-group20-ReverseComplementary.fasta ./nohup docker run -v /var/run/docker.sock:/var/run/docker.sock -v `pwd`:/data -w /data test_tools:47.0_genome_compare_revised python /usr/bin/genome_comepare.py mangguo-canu-hicpro-allhic-ok-group4-group9-group15-group17-group16-group20-ReverseComplementary.fasta GCF_011075055-ok.fasta &
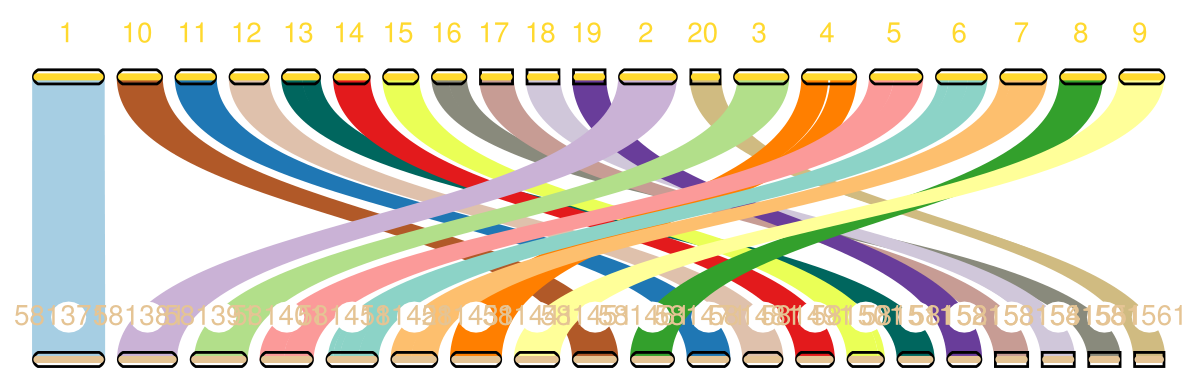
(5)将反向互补后的染色体重新进行排序并绘制基因组共线性结果。
vim seqids-two-linedocker run -v /var/run/docker.sock:/var/run/docker.sock -v `pwd`:/data -w /data genome_collinearity_plot_docker python -m jcvi.graphics.karyotype seqids-two-line layout-two-line
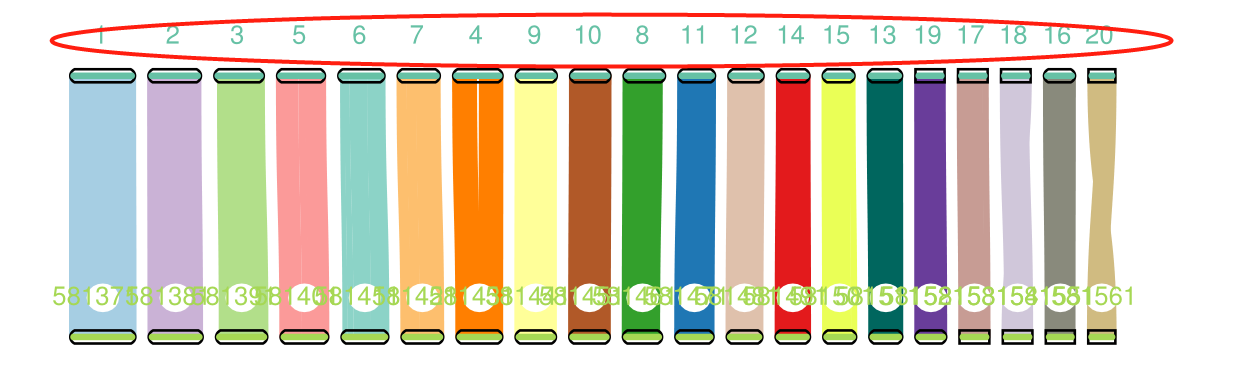
(6)将重新排序后的染色体序列ID进行重命名
将上一步1,2,3,5,6,7,8,4,9,10,8,11,12,14,15,13,19,17,18,16,20号染色体分别命名为1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,然后再进行共线性分析。python chrom-order-rename.py mangguo-canu-hicpro-allhic-ok-group4-group9-group15-group17-group16-group20-ReverseComplementary.fasta group1,group2,group3,group5,group6,group7,group8,group4,group9,group10,group8,group11,group12,group14,group15,group13,group19,group17,group18,group16,group20生成mangguo-canu-hicpro-allhic-ok-group4-group9-group15-group17-group16-group20-ReverseComplementary-reorder-chromrename.fasta新文件,这个文件中,group1被命名为chr1,group2被命名为chr2,group3被命名为chr3,group5被命名为chr4,group6被命名为chr5等等nohup docker run -v /var/run/docker.sock:/var/run/docker.sock -v `pwd`:/data -w /data test_tools:47.0_genome_compare_revised python /usr/bin/genome_comepare.py mangguo-canu-hicpro-allhic-ok-group4-group9-group15-group17-group16-group20-ReverseComplementary-reorder-chromrename.fasta GCF_011075055-ok.fasta &
import os
import sys
chrs = sys.argv[2].strip().split(",")
print(chrs)
result = sys.argv[1].strip() + "-" + str("-".join(chrs)) + "-ReversedComplementary_result"
os.system('rm -rf %s && mkdir %s '%(result,result))
os.chdir('./%s'%(result))
os.system('cp ../%s ./'%(sys.argv[1]))
all_chrs = os.popen('grep ">" %s | cut -d ">" -f 2 '%(sys.argv[1])).read().strip().split("\n")
print(all_chrs)
for ele in all_chrs :
if ele not in chrs :
with open("nochange.id","a+") as f:
f.write(ele.strip() + "\n")
else :
with open("change.id","a+") as f:
f.write(ele.strip() + "\n")
os.system('seqkit grep -f nochange.id %s | seqkit seq -w 0 - > f1.fasta '%(sys.argv[1]))
os.system('seqkit grep -f change.id %s | seqkit seq -r -p - | seqkit seq -w 0 - > f2.fasta '%(sys.argv[1]))
os.system('cat f1.fasta f2.fasta > f3.fasta')
dic_1 = {}
f1 = open("f3.fasta").read().strip().split(">")[1:]
for eles in f1 :
dic_1[eles.strip().split("\n")[0]] = eles.strip().split("\n")[1]
fo = sys.argv[1].strip().split(".fa")[0] + "-" + str("-".join(chrs)) + "-ReverseComplementary.fasta"
#os.system('cat f1.fasta f2.fasta > %s'%(fo))
for ele in all_chrs :
with open(fo,"a+") as f:
f.write(">" + ele.strip() + "\n" + dic_1[ele].strip() + "\n" )
chrom-order-rename.py
import os
import sys
chrn = sys.argv[2].strip().split(",")
fo = sys.argv[1].strip().split(".fa")[0] + "-reorder-chromrename.fasta"
os.system('rm -rf tmp.id tmp.fasta %s'%(fo))
for ele in chrn :
os.system('echo "%s" > tmp.id '%(ele.strip()))
os.system('seqkit grep -f tmp.id %s -w 0 >> tmp.fasta '%(sys.argv[1] ) )
print(ele.strip())
i = 1
for ele in open("tmp.fasta").read().strip().split(">")[1::] :
with open(fo,"a+") as f :
f.write(">" + "chr" + str(i) + "\n" + ele.strip().split("\n")[1].strip() + "\n" )
#f.write(">" + "chr" + str(i) + "\n" )
i = i + 1
os.system('rm -rf tmp.id tmp.fasta ')


