R语言编程
20231005_ClusterGVis按时间序列对基因表达进行聚类及可视化
888

ClusterGVis包可以基于 Mfuzz 的 fuzzy c-means 聚类算法或者ComplexHeatmap 的 row_km 的 Kmeans 聚类算法对RNA数据进行聚类和可视化,输入数据是标准化的 tpm/fpkm/rpkm 表达矩阵。该包用于在具有时间序列特征的转录组、蛋白质组数据中分析基因或蛋白表达的时间趋势,并将具有相似表达模式的基因或蛋白划分聚类,帮助了解这些生物学分子的动态模式以及与功能的联系。
软件包安装:
# 以管理员的身份运行Rstudio
#R 4.3.1
#Rtools 4.3
#Biocmanager 3.17
#install.packages("BiocManager")
library(BiocManager)
library(devtools)
setwd("C:/Users/lenovo/Desktop/博客文章_20231004")
#安装monocle3
BiocManager::install(version = '3.17')
library(monocle3)
BiocManager::install(c('BiocGenerics', 'DelayedArray', 'DelayedMatrixStats',
'limma', 'lme4', 'S4Vectors', 'SingleCellExperiment',
'SummarizedExperiment', 'batchelor', 'HDF5Array',
'terra', 'ggrastr'))
devtools::install_github('cole-trapnell-lab/monocle3')
library(monocle3)
#BiocManager::install("org.Mm.eg.db")
library(org.Mm.eg.db)
devtools::install_local("ClusterGVis-main.zip")
devtools::install_github("junjunlab/ClusterGVis")
library(ClusterGVis)
install.packages("Mfuzz")
BiocManager::install("Mfuzz")
library(Mfuzz)
library(Biobase)
library(Mfuzz)
library(clusterProfiler)
#BiocManager::install("GO.db")
library(GO.db)
测试数据:
这个数据集exps是一个时间序列的基因表达数据集。每一列代表一个不同的时间点,每一行代表一个基因。数据中的数值代表的是在特定时间点特定基因的表达水平。
> data(exps)
> head(exps,5)
zygote t2.cell t4.cell t8.cell tmorula blastocyst
Oog4 1.3132282 1.2370781 1.325978 1.262073 0.6549312 0.2067114
Psmd9 1.0917337 1.3159888 1.174417 1.064756 0.8685598 0.4845448
Sephs2 0.9859232 1.2010257 1.123076 1.084673 0.8878931 0.7174088
Nhlrc2 0.9856354 1.0387869 1.061926 1.076825 0.9716945 0.8651322
Trappc4 1.0775310 0.9757542 1.065544 1.080973 0.9732145 0.8269832
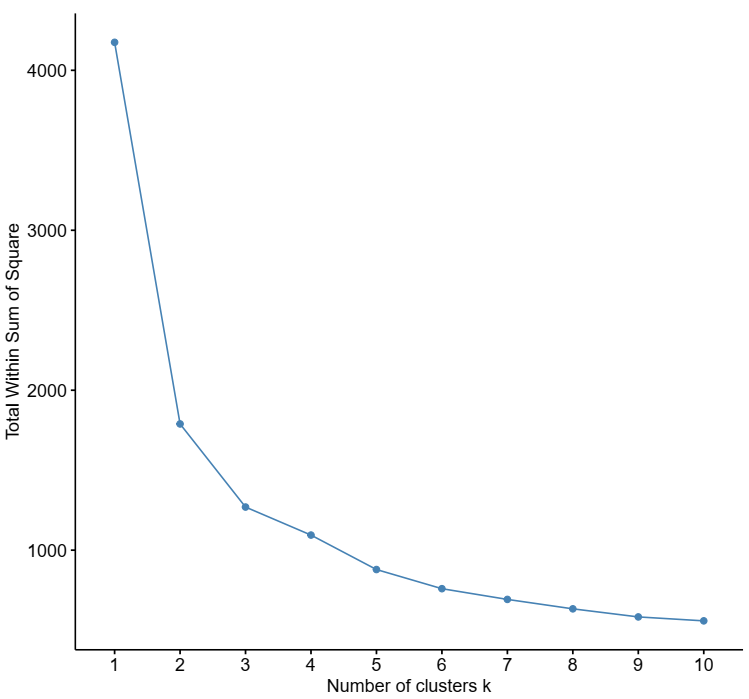
选择聚类个数:
getClusters 函数计算均方和, 可根据拐点确定最佳聚类个数
getClusters(exp = exps)
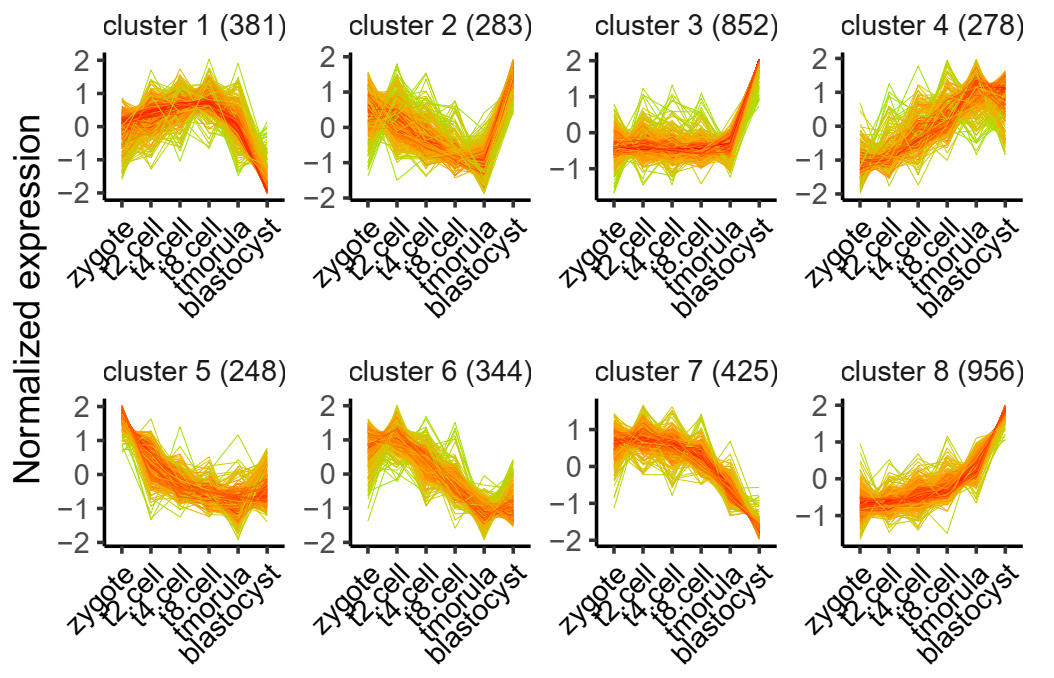
Mfuzz 聚类及可视化:
cm <- clusterData(exp = exps, cluster.method = "mfuzz", cluster.num = 8)
p2 <- visCluster(object = cm,
plot.type = "line",
ms.col = c("green","orange","red"),
add.mline = FALSE)
ggsave("mfuzz.pdf", p2, width = 7, height = 4, dpi=300)
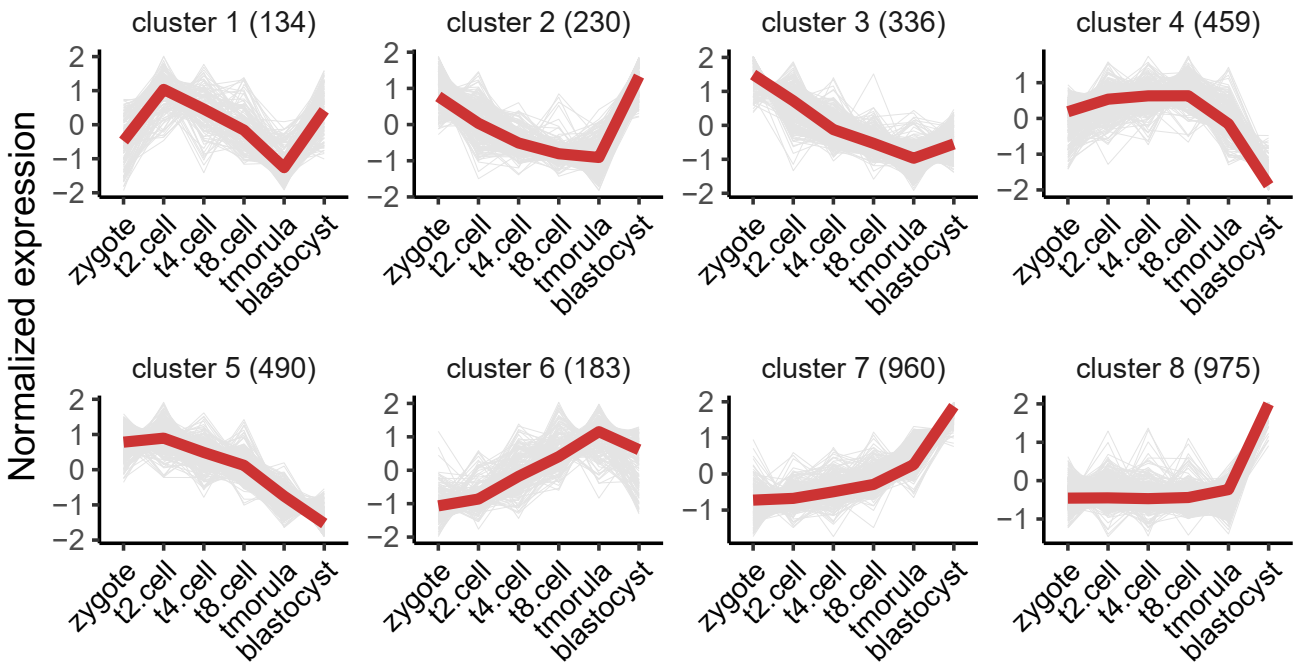
K-means聚类及可视化:
# using complexheatmap row_km for clustering
# kmeans
ck <- clusterData(exp = exps, cluster.method = "kmeans", cluster.num = 8)
# plot line only with kmeans method
p3 <- visCluster(object = ck, plot.type = "line")
ggsave("kmeans.pdf", p3, width = 7, height = 4, dpi=300)
热图+折线图:
pdf('testHT.pdf',height = 9.5,width = 10.5)
visCluster(object = ck,
plot.type = "both",
column_names_rot = 45)
dev.off()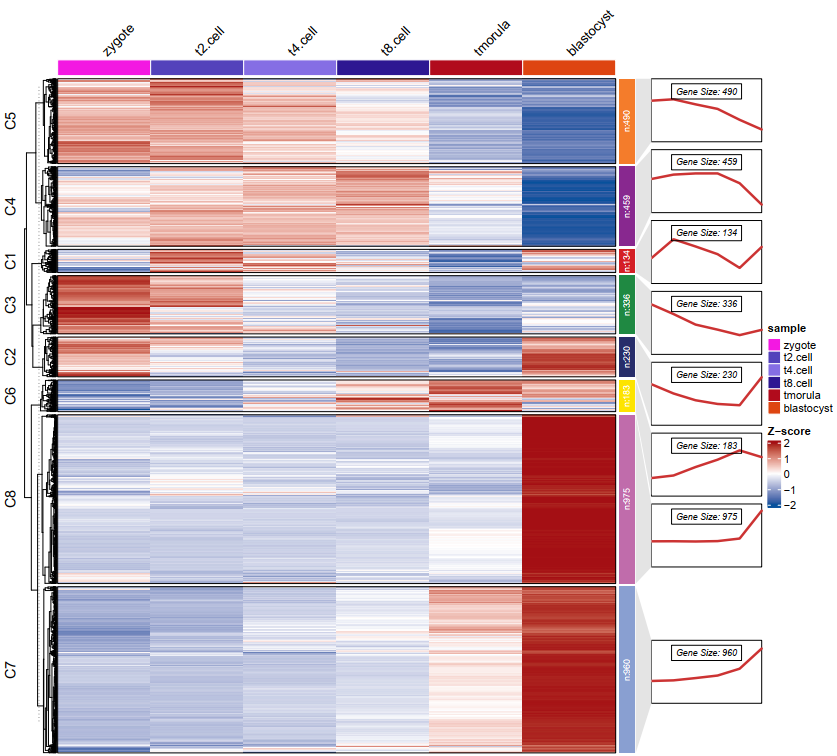
热图+折线图+GO富集结果
# load term info
data("termanno")
> head(termanno,4)
id term
1 C1 developmental process
2 C1 anatomical structure development
3 C1 multicellular organism development
4 C2 system development
# anno with GO terms
pdf('testHTterm.pdf',height = 10,width = 13)
visCluster(object = ck,
plot.type = "both",
column_names_rot = 45,
annoTerm.data = termanno)
dev.off()
# remove tree
pdf('testHTtermCmlsrt.pdf',height = 10,width = 10)
visCluster(object = cm,
plot.type = "both",
column_names_rot = 45,
annoTerm.data = termanno,
line.side = "left",
show_row_dend = F)
dev.off()

软件安装遇到的报错及解决办法:
#报错 1
package ‘sjlabelled’ successfully unpacked and MD5 sums checked
Error: Failed to install 'ClusterGVis-main.zip' from local:
Failed to install 'jjAnno' from GitHub:
ERROR: failed to lock directory ‘E:\softwares\R\R-4.0.3\library’ for modifying
Try removing ‘E:\softwares\R\R-4.0.3\library/00LOCK’这个报错如何解决
#解决办法
unlink("E:/softwares/R/R-4.0.3/library/00LOCK", recursive = TRUE)
#报错2
cm <- clusterData(exp = exps, cluster.method = "mfuzz", cluster.num = 4)
Error in (function (classes, fdef, mtable) :
函数‘exprs’标签‘"ExpressionSet"’找不到继承方法
#解决办法
退出R,然后以管理员身份重新进入Rstudio,然后仅加载library(ClusterGVis)这个包即可,上述的报错是因为加载的包种类太多,导致程序出现选择算法出现错误。
#报错 3
#devtools::install_github("junjunlab/ClusterGVis")
#Error: Failed to install 'unknown package' from GitHub:
# HTTP error 403.
#API rate limit exceeded for 114.247.56.158. (But here's the good news: Authenticated requests get a higher rate limit. Check out the documentation for more details.)
#Rate limit remaining: 0/60
#Rate limit reset at: 2023-10-04 07:06:23 UTC
#To increase your GitHub API rate limit
#Use usethis::create_github_token() to create a Personal Access Token.
#Use usethis::edit_r_environ() and add the token as GITHUB_PAT.这个报错如何解决
#解决办法
#https://poe.com/s/E46Is9u9r2cDWOMbSwuc
#usethis::edit_r_environ()
你的报错信息指出你已经达到了 GitHub API 的请求限制。GitHub 为了防止滥用,对每个 IP 地址的请求次数有一定的限制。如果你在一定时间内的请求次数超过这个限制,GitHub 就会拒绝你的请求。不过,你可以通过认证的方式来获取更高的请求限制。
下面是解决这个问题的步骤:
创建一个 GitHub 个人访问令牌(Personal Access Token, PAT):
登录你的 GitHub 账户。
点击你的头像,然后选择 "Settings"。
在左侧栏中,选择 "Developer settings"。
然后选择 "Personal access tokens"。
点击 "Generate new token"。
在 "Note" 区域输入一个描述,然后在 "Select scopes" 区域选择你需要的权限。如果你只是需要安装 R 包,你可以不选择任何权限。
点击页面底部的 "Generate token"。
将生成的令牌复制并保存好,因为你之后将无法再次查看这个令牌。
在 R 中使用你的 GitHub 个人访问令牌:
你可以使用 usethis::edit_r_environ() 函数在你的环境变量中存储你的 GitHub 个人访问令牌。这样,在你之后的请求中,devtools::install_github() 就会自动使用这个令牌。
运行下面的代码:
usethis::edit_r_environ()
这将打开一个文件,你可以在其中添加一行:
GITHUB_PAT=你的令牌
GITHUB_PAT=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
然后保存并关闭文件。你需要重启 R 来使这个变动生效。
标签:
rstudio


北京 天气
晴
-7℃
最新文章
推荐阅读
网站浏览
